Supplemental Figure 1
Paul Hook
Last update: 2018-01-18
Code version: 5996c19850dc5fc65723f30bc33ab1c8d083a7e6
Setting important directories. Also loading important libraries and custom functions for analysis.
seq_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/data"
file_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/output"
Rdata_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/data"
Script_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/code"
source(file.path(Script_dir,'init.R'))
source(file.path(Script_dir,"tools_R.r"))
#loading any special libraries
library(readr)
library(cowplot)Loading the data needed
The final ‘dat’ cds was loaded and some summary stats were calculated to quickly check the data. Also the PCA plots from P7 regions were loaded in.
# Filtered final cds
dat.filter <- readRDS(file.path(Rdata_dir,"dat.filter.final.Rds"))
nrow(pData(dat.filter)) # 396## [1] 396with(pData(dat.filter), table(age, region))## region
## age FB MB OB
## E15.5 84 88 0
## P7 81 75 68# Outlier plots
FB.outlier.plot <- readRDS(file = file.path(Rdata_dir,"P7.FB.PCA.outliers.rds"))
MB.outlier.plot <- readRDS(file = file.path(Rdata_dir,"P7.MB.PCA.outliers.rds"))
OB.outlier.plot <- readRDS(file = file.path(Rdata_dir,"P7.OB.PCA.outliers.rds"))Figure S1A
Histogram displaying number of genes per cell in the final filtered dataset.
# Number of genes per cell
max.expressed <- round(max(pData(dat.filter)$num_genes_expressed))
min.expressed <- round(min(pData(dat.filter)$num_genes_expressed))
mean.expressed <- round(mean(pData(dat.filter)$num_genes_expressed))
# Writing out histogram to pdf
pdf(file = file.path(file_dir, "Figure.S1A.pdf"), width = 6, height = 6)
hist(pData(dat.filter)$num_genes_expressed,
breaks=25,
main = NA,
col="steelblue",
xlab = "Number of genes expressed",
ylab = "Number of cells",
ylim = c(0,30))
mtext(text = paste("Min =",min.expressed), adj = 0.15, padj = 10)
mtext(text = paste("Max =", max.expressed), adj = 0.15, padj = 12)
mtext(text = paste("Mean =",mean.expressed), adj = 0.15, padj = 14)
title("Genes expressed per cell", line = -0.3)
dev.off()## quartz_off_screen
## 2# Plotting histogram
hist(pData(dat.filter)$num_genes_expressed,
breaks=25,
main = NA,
col="steelblue",
xlab = "Number of genes expressed",
ylab = "Number of cells",
ylim = c(0,30))
mtext(text = paste("Min =",min.expressed), adj = 0.15, padj = 10)
mtext(text = paste("Max =", max.expressed), adj = 0.15, padj = 12)
mtext(text = paste("Mean =",mean.expressed), adj = 0.15, padj = 14)
title("Genes expressed per cell", line = -0.3)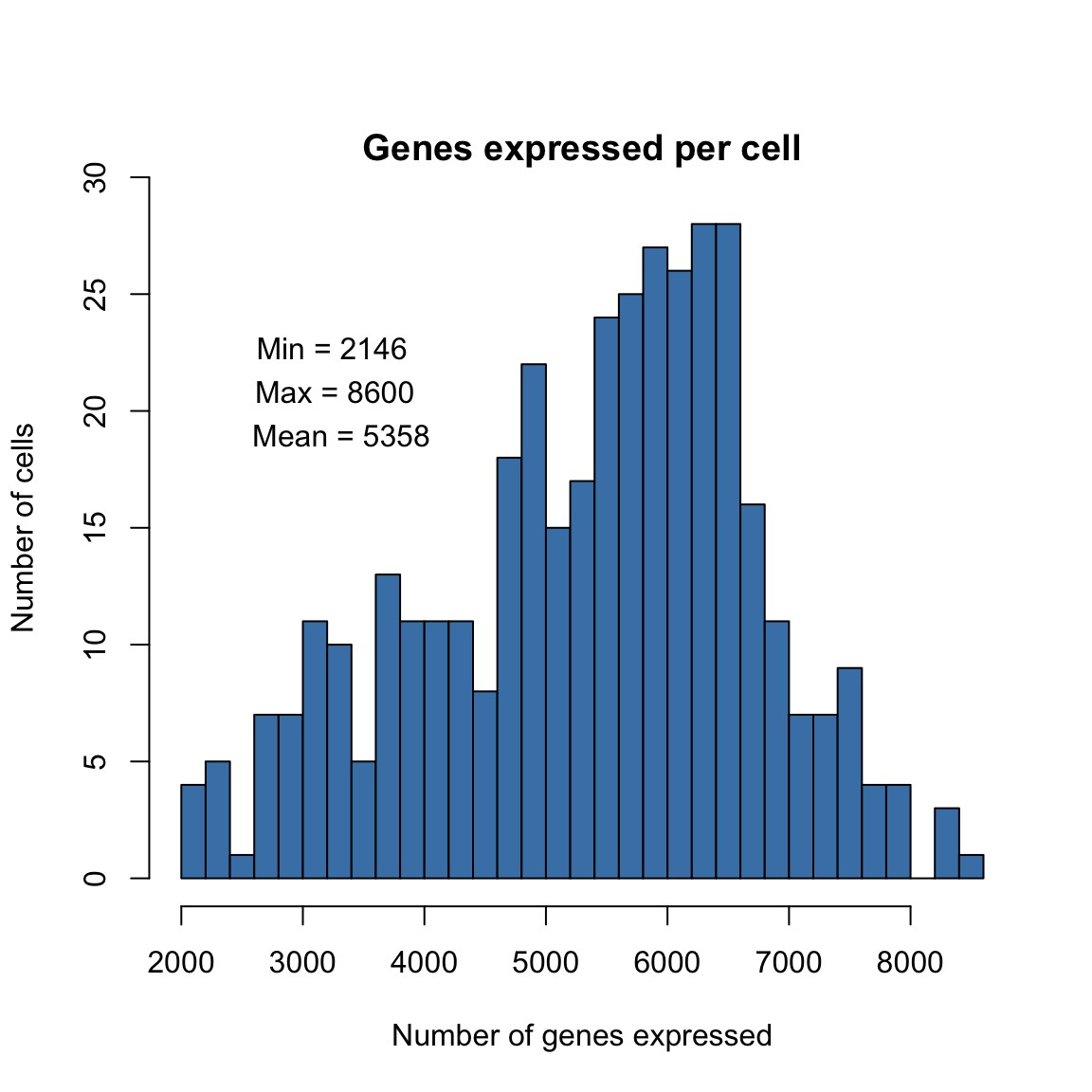
Figure S1B
Histogram displaying mRNA per cell in the final filtered dataset.
# mRNA per cell
max.mRNAs <- round(max(pData(dat.filter)$Total_mRNAs))
min.mRNAs <- round(min(pData(dat.filter)$Total_mRNAs))
mean.mRNAs <- round(mean(pData(dat.filter)$Total_mRNAs))
# Writing out histograms to pdf
pdf(file = file.path(file_dir, "Figure.S1B.pdf"), width = 6, height = 6)
hist(pData(dat.filter)$Total_mRNAs,breaks=50,
col="darkgreen",
main = NA,
ylab = "Number of cells", xlab = "Total mRNAs",
ylim = c(0,25))
mtext(text = paste("Min =",min.mRNAs), adj = 0.15, padj = 10)
mtext(text = paste("Max =", max.mRNAs), adj = 0.15, padj = 12)
mtext(text = paste("Mean =",mean.mRNAs), adj = 0.15, padj = 14)
title("Total mRNA per cell", line = -0.3)
dev.off()## quartz_off_screen
## 2# Plotting the histogram
hist(pData(dat.filter)$Total_mRNAs,breaks=50,
col="darkgreen",
main = NA,
ylab = "Number of cells", xlab = "Total mRNAs",
ylim = c(0,25))
mtext(text = paste("Min =",min.mRNAs), adj = 0.15, padj = 10)
mtext(text = paste("Max =", max.mRNAs), adj = 0.15, padj = 12)
mtext(text = paste("Mean =",mean.mRNAs), adj = 0.15, padj = 14)
title("Total mRNA per cell", line = -0.3)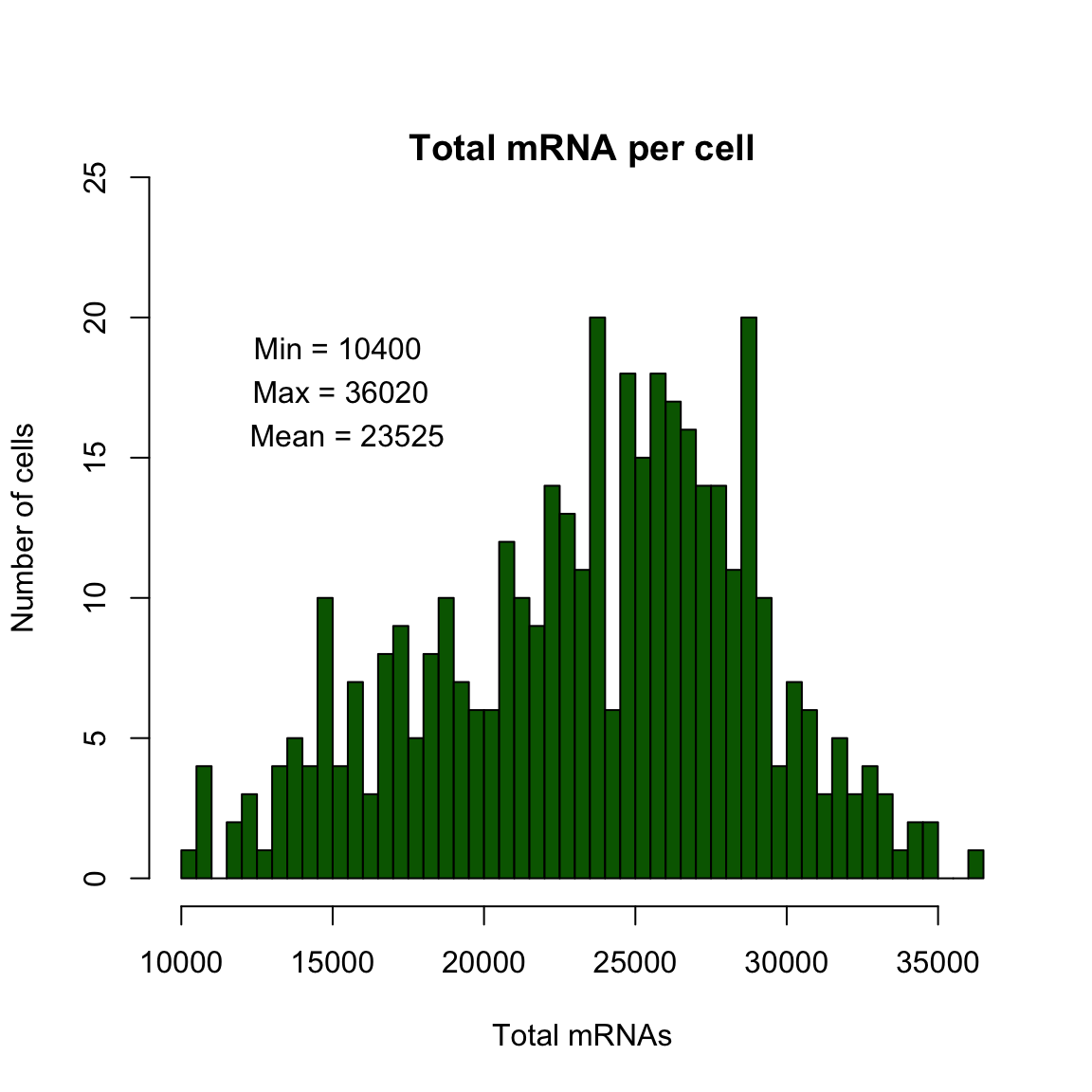
Figure S1C
Histogram displaying mass per cell in the final filtered dataset.
# Mass per cell
max.mass <- round(max(pData(dat.filter)$total_mass))
min.mass <- round(min(pData(dat.filter)$total_mass))
mean.mass <- round(mean(pData(dat.filter)$total_mass))
# Writing out the histogram to a PDF
pdf(file = file.path(file_dir, "Figure.S1C.pdf"), width = 6, height = 6)
hist(pData(dat.filter)$total_mass,
breaks=50,
main = NA,
col="darkred",
ylab = "Number of cells", xlab = "Total mass\n(fragments mapped to transcriptome)",
ylim = c(0,20))
mtext(text = paste("Min =",min.mass), adj = 0.85, padj = 10)
mtext(text = paste("Max =", max.mass), adj = 0.85, padj = 12)
mtext(text = paste("Mean =",mean.mass), adj = 0.85, padj = 14)
title("Total mass per cell", line = -0.3)
dev.off()## quartz_off_screen
## 2# Plotting the histogram
hist(pData(dat.filter)$total_mass,
breaks=50,
main = NA,
col="darkred",
ylab = "Number of cells", xlab = "Total mass\n(fragments mapped to transcriptome)",
ylim = c(0,20))
mtext(text = paste("Min =",min.mass), adj = 0.85, padj = 10)
mtext(text = paste("Max =", max.mass), adj = 0.85, padj = 12)
mtext(text = paste("Mean =",mean.mass), adj = 0.85, padj = 14)
title("Total mass per cell", line = -0.3)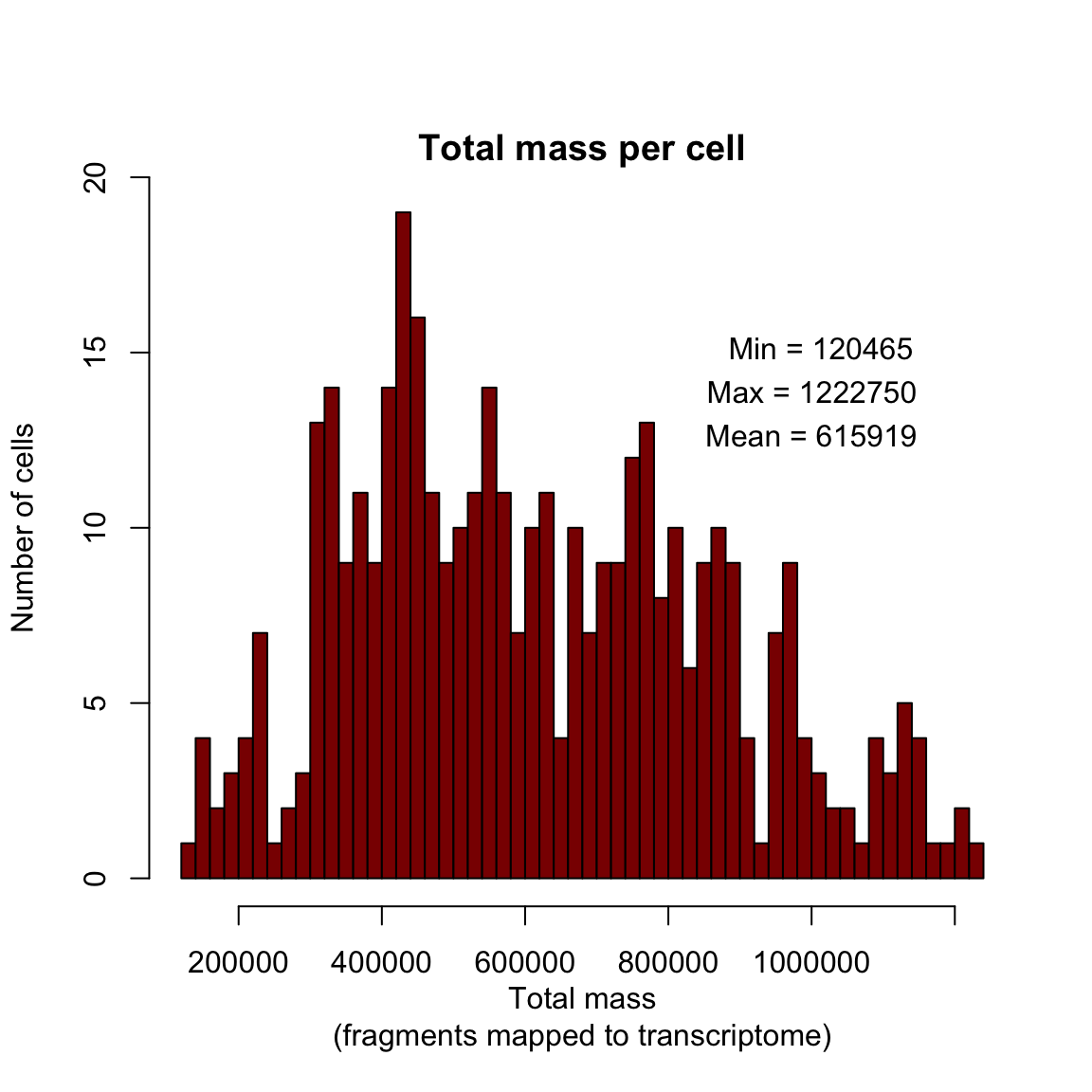
Figure S1D
Barplots showing the number of final cells by condition assayed after filtering
# Number of cells x condition
p <- ggplot(pData(dat.filter))
q <- p + geom_bar(aes(x=region,fill=region),width=0.5) + ggtitle("Valid cells per condition") + theme_bw() + scale_fill_brewer(palette="Set1", guide = F) + facet_grid(.~age) + ylab("Number of cells") + xlab("Region") + theme(plot.title = element_text(hjust = 0.5))
pdf(file = file.path(file_dir, "Figure.S1D.pdf"), width = 6, height = 4)
q
dev.off()## quartz_off_screen
## 2q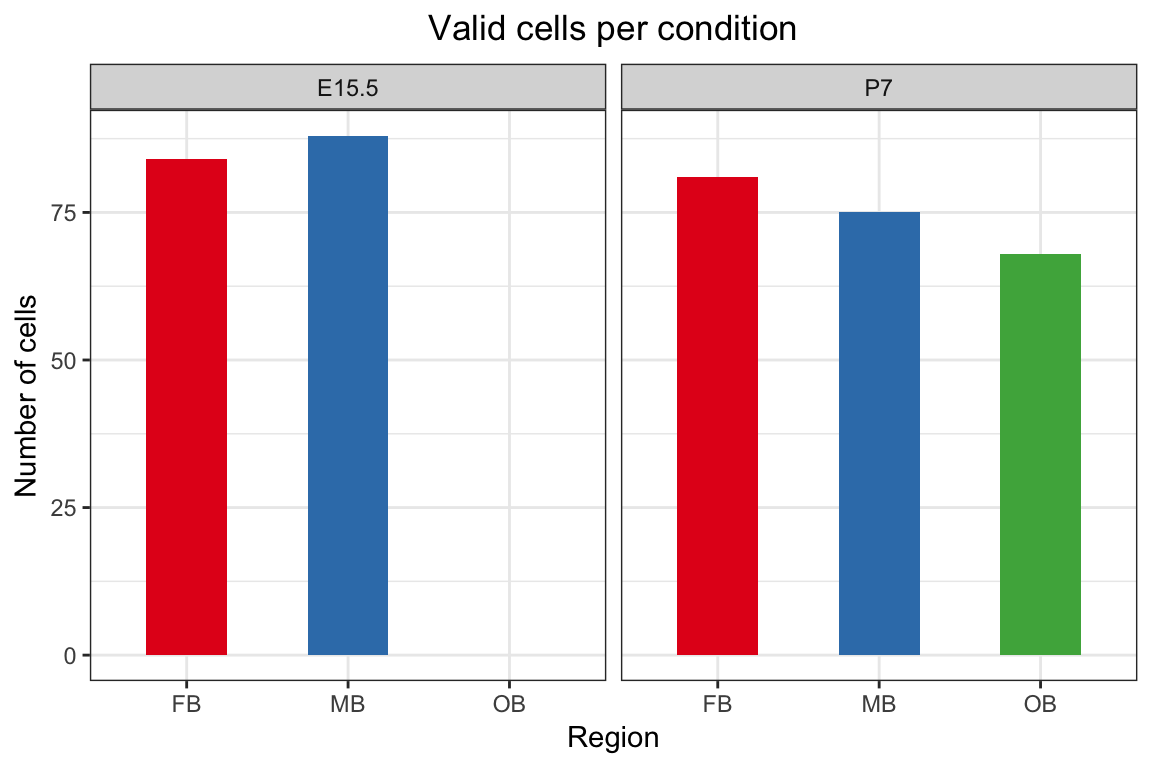
Figure S1E
PCA plots displaying outliers that were removed during data filtering. Note that circles appearing on the plots were removed.
fb <- FB.outlier.plot + scale_color_manual(values = brewer.pal(8,"Set1")[1], name = "Region") + monocle:::monocle_theme_opts() + ggtitle("P7 FB PCA (PC1 vs. PC2)") +
theme(plot.title = element_text(hjust = 0.5),
axis.title.y = element_text(family = 'Helvetica',size=10),
axis.title.x = element_text(family = 'Helvetica',size=10))
mb <- MB.outlier.plot + scale_color_manual(values = brewer.pal(8,"Set1")[2], name = "Region") + monocle:::monocle_theme_opts() + ggtitle("P7 MB PCA (PC1 vs. PC2)") +
theme(plot.title = element_text(hjust = 0.5),
axis.title.y = element_text(family = 'Helvetica',size=10),
axis.title.x = element_text(family = 'Helvetica',size=10))## Scale for 'colour' is already present. Adding another scale for
## 'colour', which will replace the existing scale.ob <- OB.outlier.plot + scale_color_manual(values = brewer.pal(8,"Set1")[3], name = "Region") + monocle:::monocle_theme_opts() + ggtitle("P7 OB PCA (PC1 vs. PC2)") +
theme(plot.title = element_text(hjust = 0.5),
axis.title.y = element_text(family = 'Helvetica',size=10),
axis.title.x = element_text(family = 'Helvetica',size=10))## Scale for 'colour' is already present. Adding another scale for
## 'colour', which will replace the existing scale.pdf(file = file.path(file_dir, "Figure.S1E.pdf"), width = 4, height = 3)
fb
ob
mb
dev.off()## quartz_off_screen
## 2plot_grid(fb,ob,mb,nrow = 1)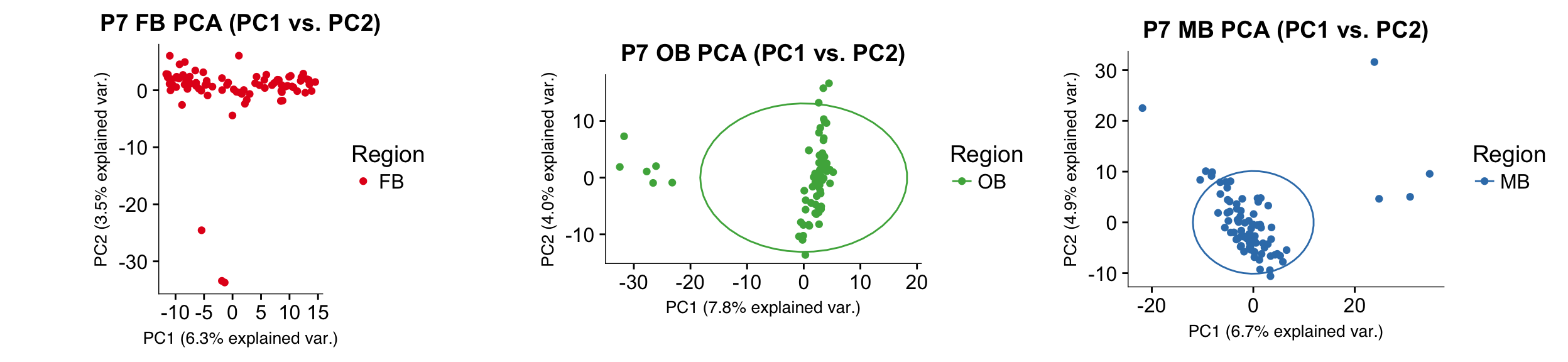
Session Info
sessionInfo()## R version 3.3.0 (2016-05-03)
## Platform: x86_64-apple-darwin13.4.0 (64-bit)
## Running under: OS X 10.11.6 (El Capitan)
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] grid splines stats4 parallel stats graphics grDevices
## [8] utils datasets methods base
##
## other attached packages:
## [1] cowplot_0.9.2 readr_1.1.1 ggbiplot_0.55
## [4] scales_0.5.0 SC3_1.1.4 ROCR_1.0-7
## [7] jackstraw_1.1.1 lfa_1.2.2 tsne_0.1-3
## [10] gridExtra_2.3 slackr_1.4.2 vegan_2.4-4
## [13] permute_0.9-4 MASS_7.3-47 gplots_3.0.1
## [16] RColorBrewer_1.1-2 Hmisc_4.0-3 Formula_1.2-2
## [19] survival_2.41-3 lattice_0.20-35 Heatplus_2.18.0
## [22] Rtsne_0.13 pheatmap_1.0.8 tidyr_0.7.1
## [25] dplyr_0.7.4 plyr_1.8.4 heatmap.plus_1.3
## [28] stringr_1.2.0 marray_1.50.0 limma_3.28.21
## [31] reshape2_1.4.3 monocle_2.2.0 DDRTree_0.1.5
## [34] irlba_2.2.1 VGAM_1.0-2 ggplot2_2.2.1
## [37] Biobase_2.32.0 BiocGenerics_0.18.0 Matrix_1.2-11
##
## loaded via a namespace (and not attached):
## [1] RSelenium_1.7.1 colorspace_1.3-2 class_7.3-14
## [4] rprojroot_1.2 htmlTable_1.9 corpcor_1.6.9
## [7] base64enc_0.1-3 mvtnorm_1.0-6 codetools_0.2-15
## [10] doParallel_1.0.11 robustbase_0.92-7 knitr_1.17
## [13] jsonlite_1.5 cluster_2.0.6 semver_0.2.0
## [16] shiny_1.0.5 rrcov_1.4-3 httr_1.3.1
## [19] backports_1.1.1 assertthat_0.2.0 lazyeval_0.2.1
## [22] acepack_1.4.1 htmltools_0.3.6 tools_3.3.0
## [25] bindrcpp_0.2 igraph_1.1.2 gtable_0.2.0
## [28] glue_1.1.1 binman_0.1.0 doRNG_1.6.6
## [31] Rcpp_0.12.14 slam_0.1-37 gdata_2.18.0
## [34] nlme_3.1-131 iterators_1.0.8 mime_0.5
## [37] rngtools_1.2.4 gtools_3.5.0 WriteXLS_4.0.0
## [40] XML_3.98-1.9 DEoptimR_1.0-8 hms_0.3
## [43] yaml_2.1.15 pkgmaker_0.22 rpart_4.1-11
## [46] fastICA_1.2-1 latticeExtra_0.6-28 stringi_1.1.5
## [49] pcaPP_1.9-72 foreach_1.4.3 e1071_1.6-8
## [52] checkmate_1.8.4 caTools_1.17.1 rlang_0.1.6
## [55] pkgconfig_2.0.1 matrixStats_0.52.2 bitops_1.0-6
## [58] qlcMatrix_0.9.5 evaluate_0.10.1 purrr_0.2.4
## [61] bindr_0.1 labeling_0.3 htmlwidgets_0.9
## [64] magrittr_1.5 R6_2.2.2 combinat_0.0-8
## [67] wdman_0.2.2 foreign_0.8-69 mgcv_1.8-22
## [70] nnet_7.3-12 tibble_1.3.4 KernSmooth_2.23-15
## [73] rmarkdown_1.8 data.table_1.10.4 HSMMSingleCell_0.106.2
## [76] digest_0.6.12 xtable_1.8-2 httpuv_1.3.5
## [79] openssl_0.9.7 munsell_0.4.3 registry_0.3This R Markdown site was created with workflowr