E15.5 MB Recursive Analysis
Paul Hook
Last update: 2018-01-18
Code version: 5996c19850dc5fc65723f30bc33ab1c8d083a7e6
Setting important directories.
Also loading important libraries and custom functions for analysis.
seq_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/data"
file_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/output"
Rdata_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/data"
Script_dir <- "/Volumes/PAULHOOK/sc-da-parkinsons/code"
source(file.path(Script_dir,'init.R'))
source(file.path(Script_dir,"tools_R.r"))Loading the E15 cds
e15.Mb.dat.filter <- readRDS(file = file.path(Rdata_dir, "e15.Mb.dat.filter.rds"))Filter genes by percentage of cells expresssing each gene
# Plot number of cells expressing each gene as histogram
hist(fData(e15.Mb.dat.filter)$num_cells_expressed,breaks=50,col="red",main="Cells expressed per gene")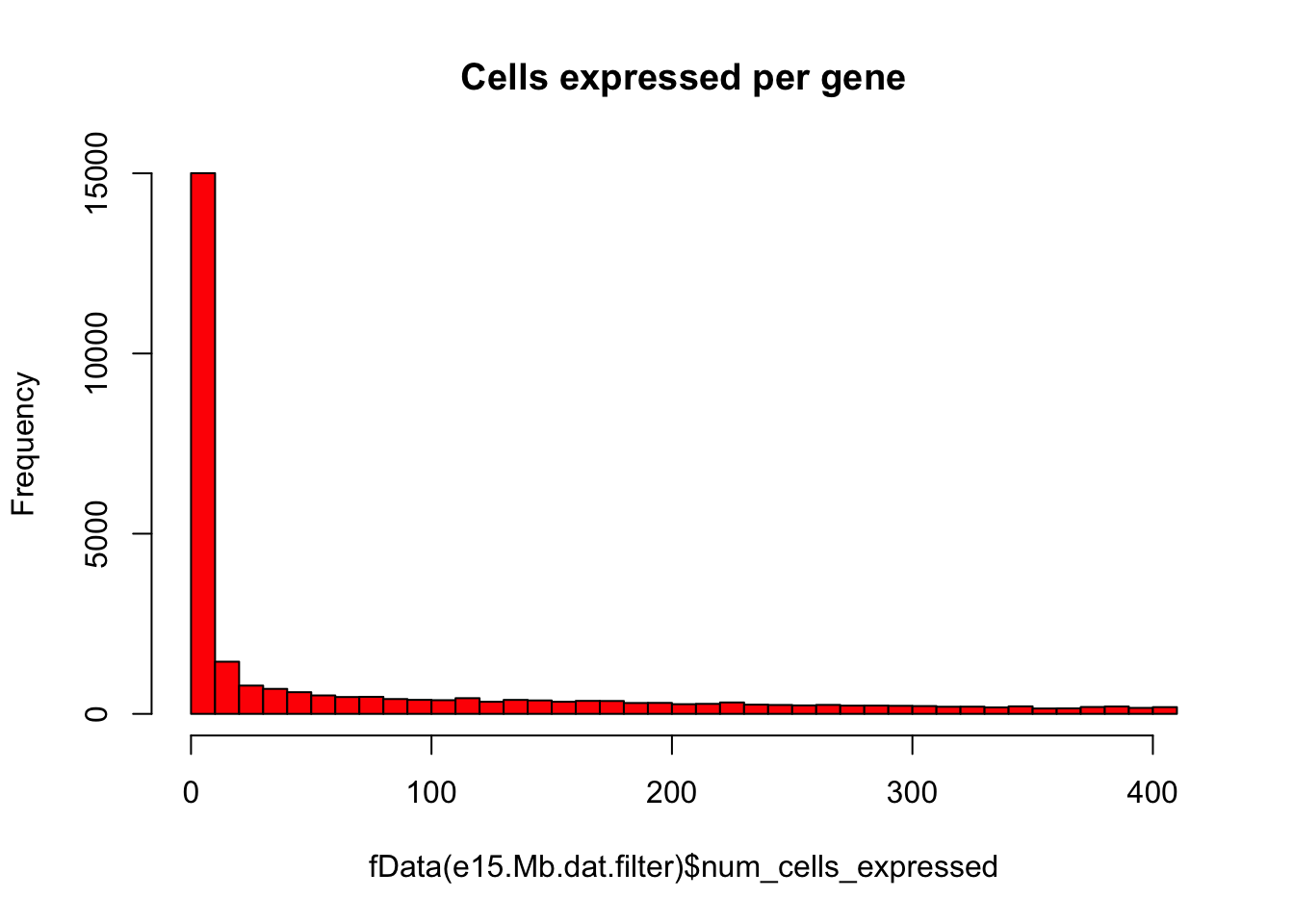
# Keep only expressed genes with expression in >= 5% of cells
numCellThreshold<-nrow(pData(e15.Mb.dat.filter))*0.05
e15.Mb.dat.expressed_genes<-row.names(subset(fData(e15.Mb.dat.filter),num_cells_expressed >= numCellThreshold))
# Same plot as above with threshold
hist(fData(e15.Mb.dat.filter)$num_cells_expressed,breaks=50,col="red",main="Cells expressed per gene - threshold")
abline(v=numCellThreshold,lty="dashed")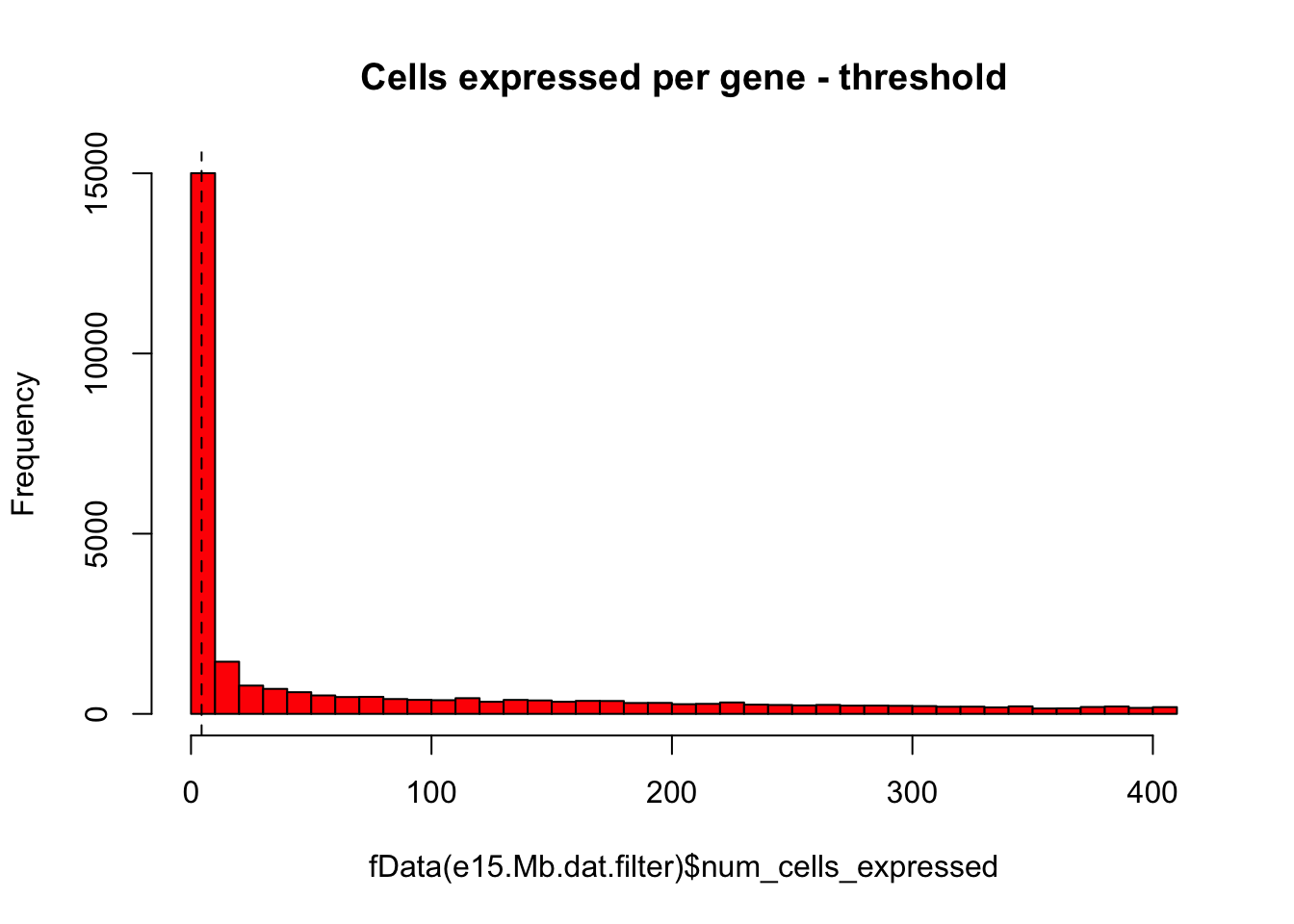
Prepping the Monocle model for analysis
# Only keeping "expressed" genes
e15.Mb.dat.filter <-e15.Mb.dat.filter[e15.Mb.dat.expressed_genes,]
# Estimating the size factors
e15.Mb.dat.filter <-estimateSizeFactors(e15.Mb.dat.filter)
# Estimating dispersions
e15.Mb.dat.filter <- estimateDispersions(e15.Mb.dat.filter,cores=8)## Warning: Deprecated, use tibble::rownames_to_column() instead.## Removing 134 outliers# Removing 134 outliers
# Warning message:
# Deprecated, use tibble::rownames_to_column() instead. Calculating summary stats
# Calculating summary stats
fData(e15.Mb.dat.filter)$mean_expr<-apply(round(exprs(e15.Mb.dat.filter)),1,mean) # mean expression
fData(e15.Mb.dat.filter)$sd_expr<-apply(round(exprs(e15.Mb.dat.filter)),1,sd) # sd expression
fData(e15.Mb.dat.filter)$bcv<-(fData(e15.Mb.dat.filter)$sd_expr/fData(e15.Mb.dat.filter)$mean_expr)**2 # calculating biological coefficient of variation
fData(e15.Mb.dat.filter)$percent_detection<-(fData(e15.Mb.dat.filter)$num_cells_expressed/dim(e15.Mb.dat.filter)[2])*100 # calculating % detectionIdentifying high dispersion genes
e15.Mb.dat.filter.genes <- e15.Mb.dat.filter # spoofing the CellDataSet
disp_table <- dispersionTable(e15.Mb.dat.filter.genes) # pulling out the dispersion table
unsup_clustering_genes <-subset(disp_table, mean_expression >= 0.5 & dispersion_empirical >= 2 * dispersion_fit) # subsetting the data to pull out genes with expression above 0.5 and dispersion empirical > 2
e15.Mb.dat.high_bcv_genes<-unsup_clustering_genes$gene_id # pulling out list of genes
e15.Mb.dat.filter.order <- setOrderingFilter(e15.Mb.dat.filter, unsup_clustering_genes$gene_id)
plot_ordering_genes(e15.Mb.dat.filter.order) # plotting the dispersion and genes## Warning: Transformation introduced infinite values in continuous y-axis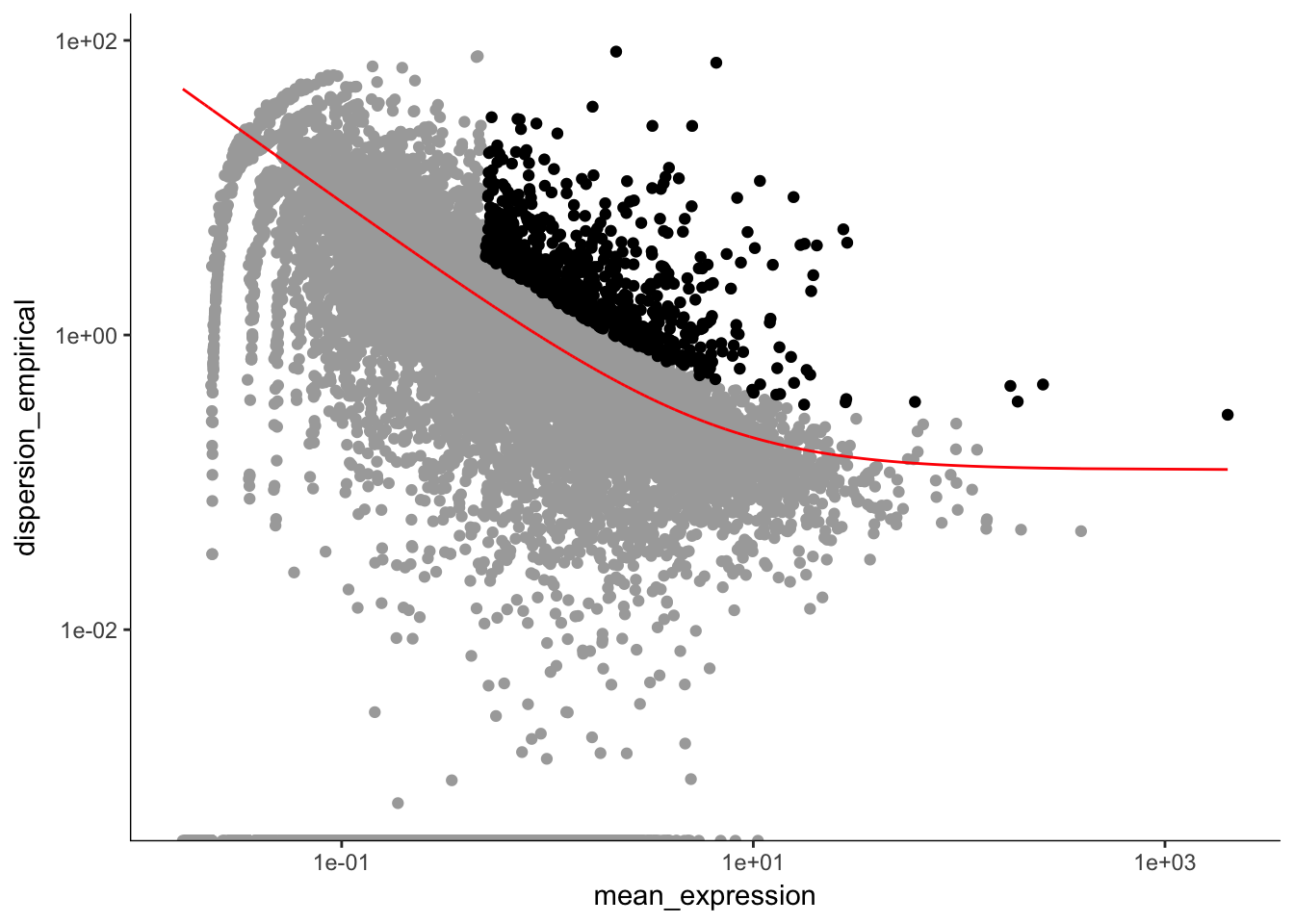
length(e15.Mb.dat.high_bcv_genes) # 778## [1] 778Running PCA with high dispersion genes
# BCV Identified high dispersion genes. Running PC analysis
e15.Mb.dat.filter.BCV.pca<-prcomp(t(log2(exprs(e15.Mb.dat.filter[e15.Mb.dat.high_bcv_genes,])+1)),center=T,scale. = TRUE)
# Plotting the PCA graphs
# Plotting the first 2 PCs and coloring by age
hvPCA1<-ggbiplot(e15.Mb.dat.filter.BCV.pca,choices=c(1,2),scale=0,groups=pData(e15.Mb.dat.filter)$age,ellipse=T,var.axes=F) + scale_color_manual(values=c("darkgreen","red")) + monocle:::monocle_theme_opts()
# Plotting the first 2 PCs and coloring by region
hvPCA2<-ggbiplot(e15.Mb.dat.filter.BCV.pca,choices=c(1,2),scale=0,groups=pData(e15.Mb.dat.filter)$region,ellipse=T,var.axes=F) + scale_color_brewer(palette="Set1") + monocle:::monocle_theme_opts()
# Plotting the first 2 PCs and coloring by plate the cell was sequenced from
hvPCA3<-ggbiplot(e15.Mb.dat.filter.BCV.pca,choices=c(1,2),scale=0,groups=pData(e15.Mb.dat.filter)$split_plate,ellipse=T,var.axes=F) + scale_color_brewer(palette="Set1") + monocle:::monocle_theme_opts()
# Show the plots in the terminal
hvPCA1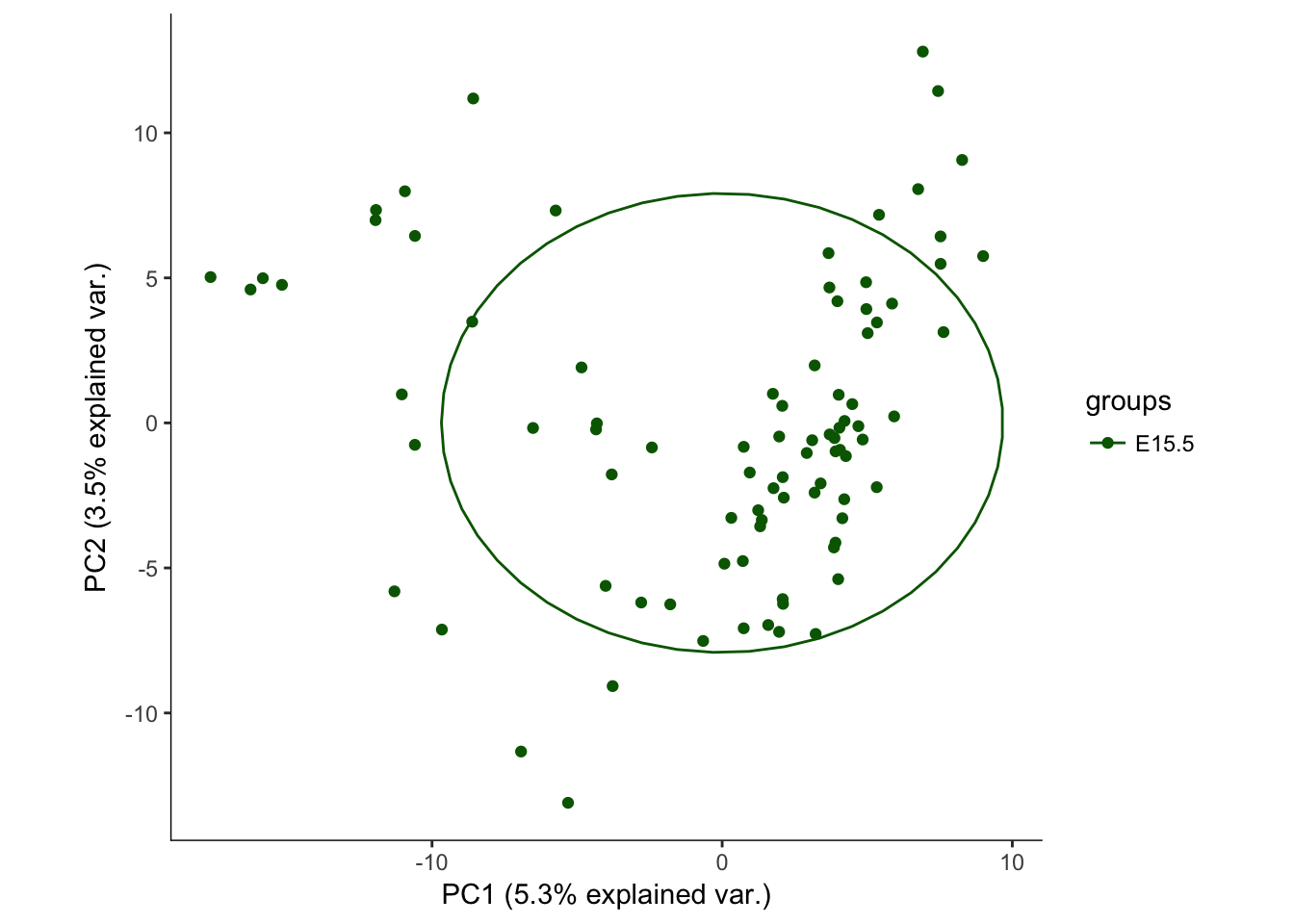
hvPCA2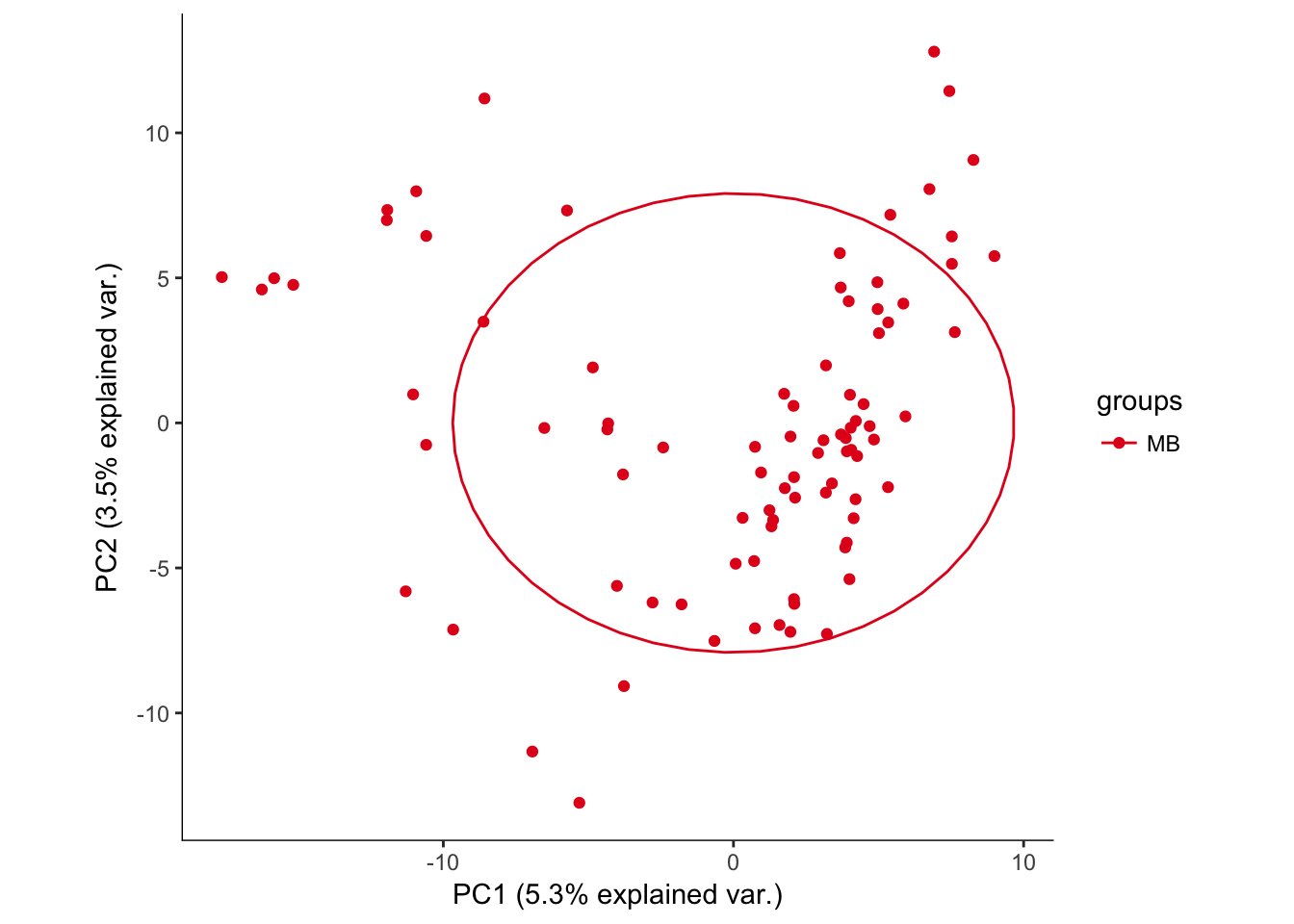
hvPCA3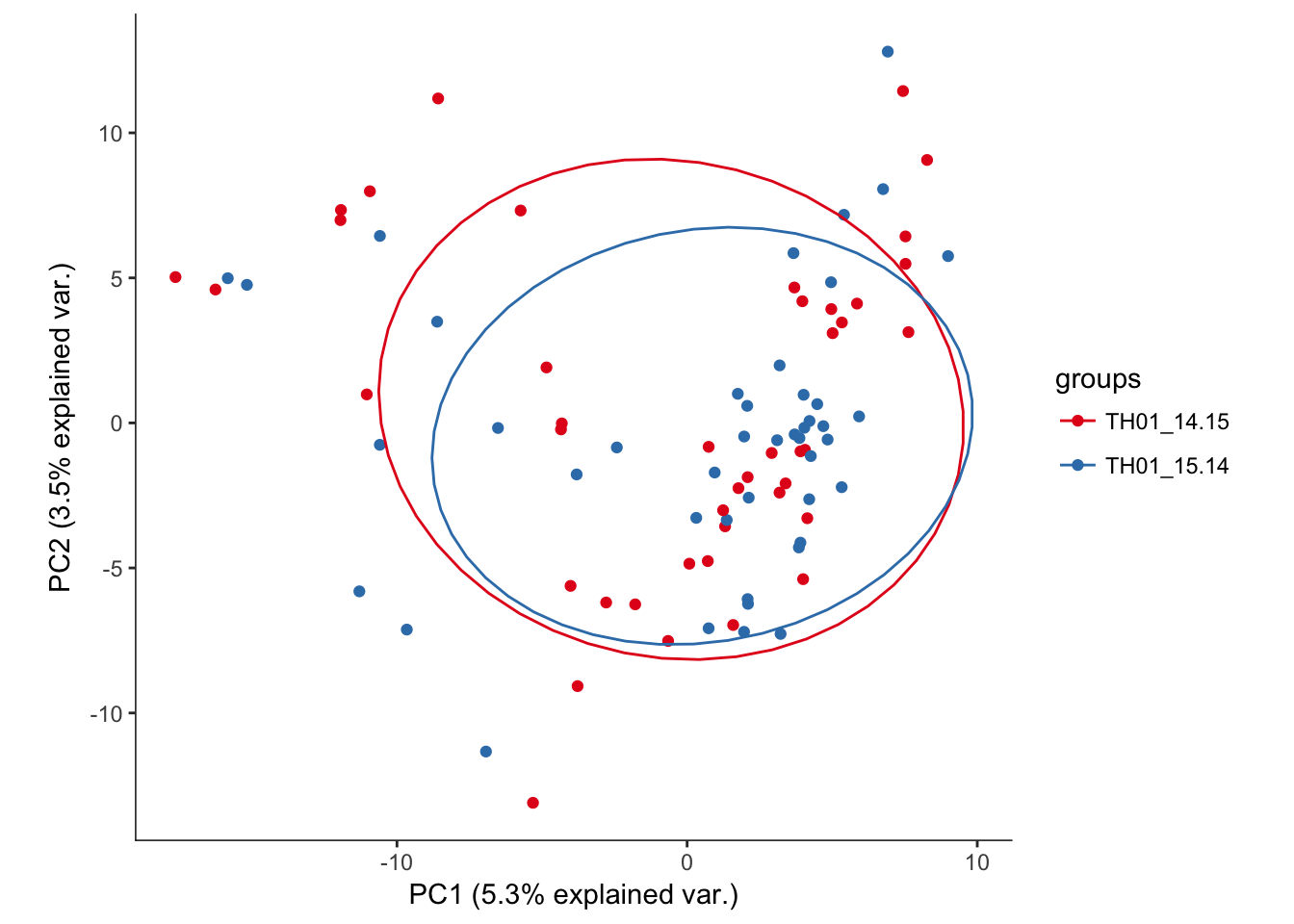
Screeplots
Viewing screeplots and determining the number of “significant” PCs
# Making a screeplot of the BCV PCA. This will help determine how many
# principal components we should use in our tSNE visualization
# Show this plot
screeplot(e15.Mb.dat.filter.BCV.pca, npcs = 30, main = "e15.5 - High BCV Genes Screeplot")
abline(v = 5, lwd = 2, col = "red")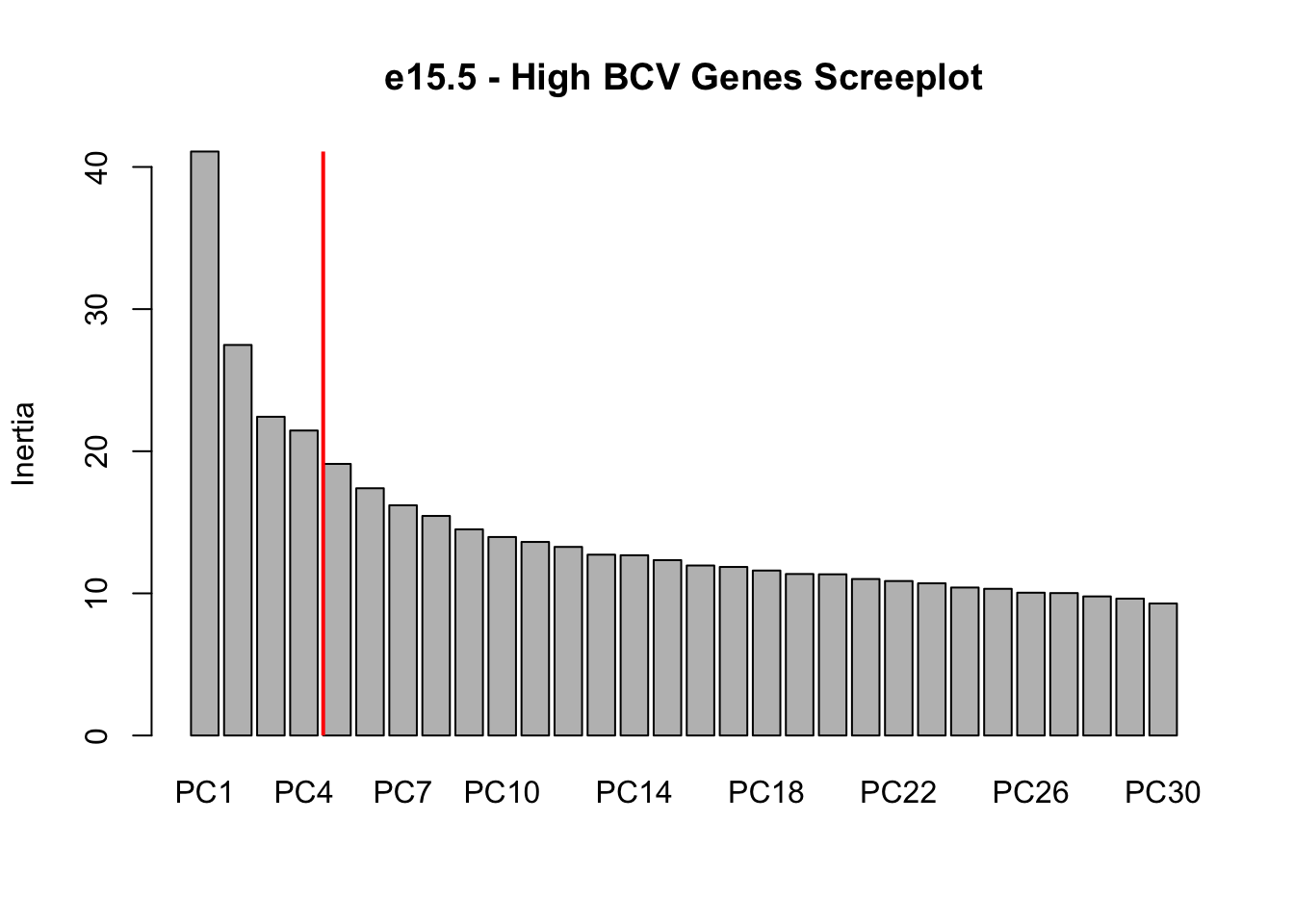
ggscreeplot(e15.Mb.dat.filter.BCV.pca)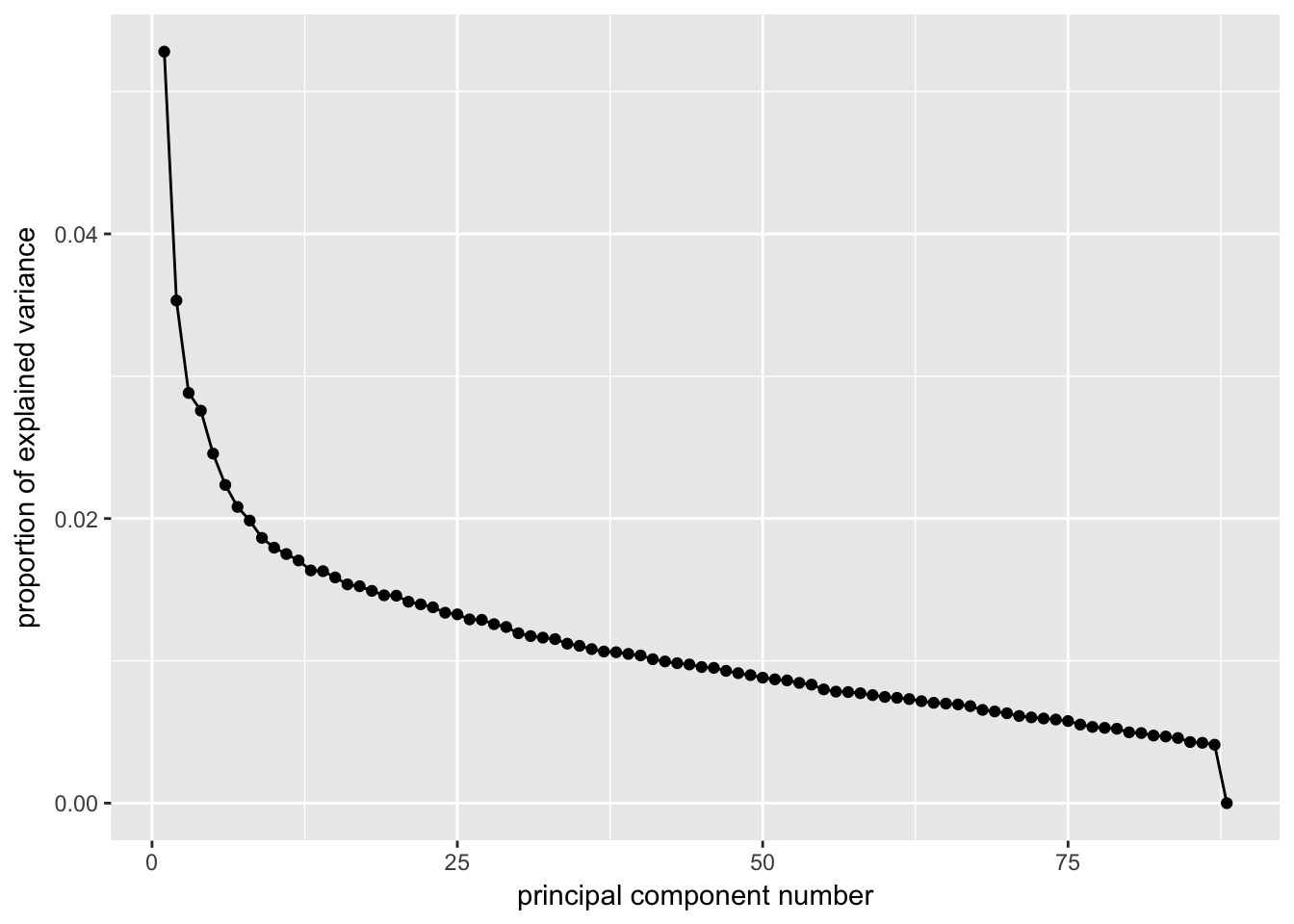
# Conclustion: Seems to be clear that just the first four PCs explain the
# most variation in our dataCreating a t-SNE plot from the “significant” PCs
nComponents<-4 # estimated from the screeplots
#seed <- runif(1,1,9999) # determined by testing random seeds
seed <- 223.3888
set.seed(seed) #setting seed
e15.Mb.dat.filter.BCV.tsne<-tsne(e15.Mb.dat.filter.BCV.pca$x[,1:nComponents],perplexity=20,max_iter=5000,whiten = FALSE)
pData(e15.Mb.dat.filter)$tSNE1_pos<-e15.Mb.dat.filter.BCV.tsne[,1]
pData(e15.Mb.dat.filter)$tSNE2_pos<-e15.Mb.dat.filter.BCV.tsne[,2]
e15.Mb.dat.filter.BCV.tsne.plot<-myTSNEPlotAlpha(e15.Mb.dat.filter,color="region", shape="age") + scale_color_brewer(palette="Set1") + ggtitle("e15.5 - BCV tSNE Plot")
e15.Mb.dat.filter.BCV.tsne.plot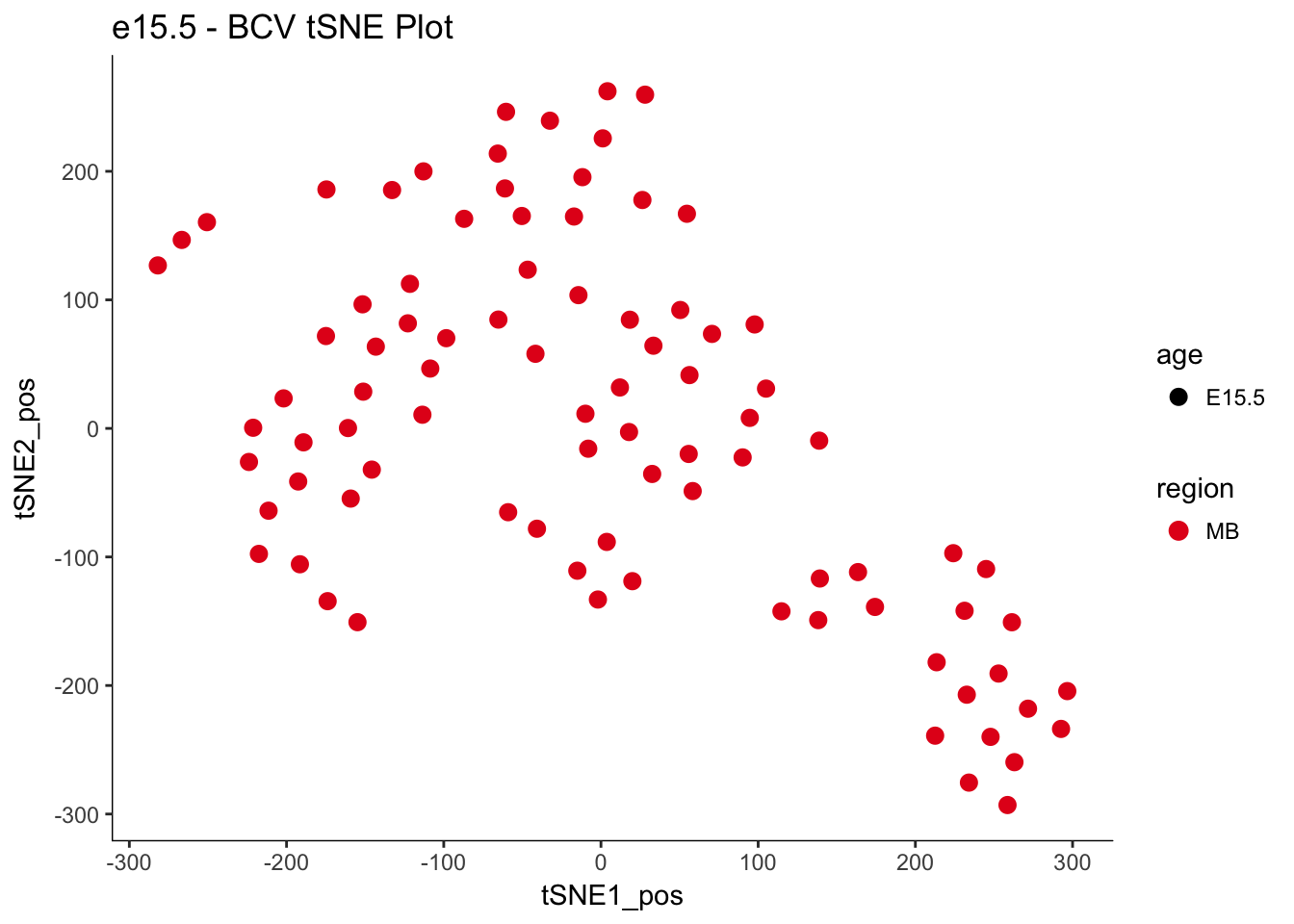
Identifying clusters
Identifying clusters in the data in an unsupervised manner
# Going to attempt to use the R program 'ADPclust' to determine how many
# clusters our data has
# Loading NbClust
library(ADPclust)
# Running ADPclust
clust.res <- adpclust(x = e15.Mb.dat.filter.BCV.tsne, draw = T)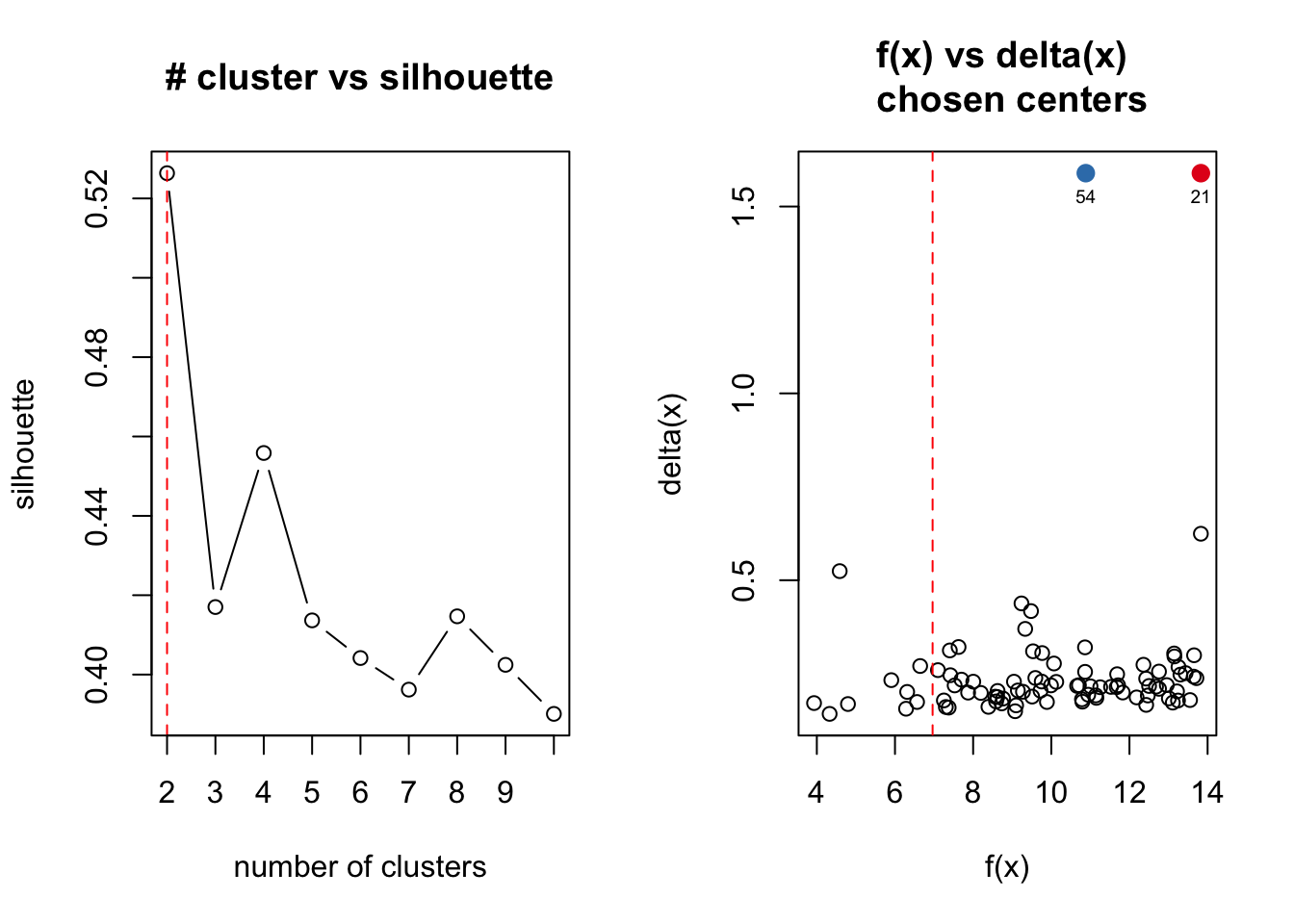
# Extracting the 'best partition' (aka the best cluster) for each cell
clust.res.df <- as.data.frame(clust.res$cluster)
# Adding the cluster assignment for each cell to the pData
pData(e15.Mb.dat.filter)$kmeans_tSNE_cluster <- as.factor(clust.res.df$`clust.res$cluster`)
# Plotting the same tSNE plot as above but coloring with the 'clusters'
myTSNEPlotAlpha(e15.Mb.dat.filter, color = "kmeans_tSNE_cluster", shape = "age") +
scale_color_brewer(palette = "Set1") + ggtitle("All Cells - BCV tSNE with Clusters Plot")## Scale for 'colour' is already present. Adding another scale for
## 'colour', which will replace the existing scale.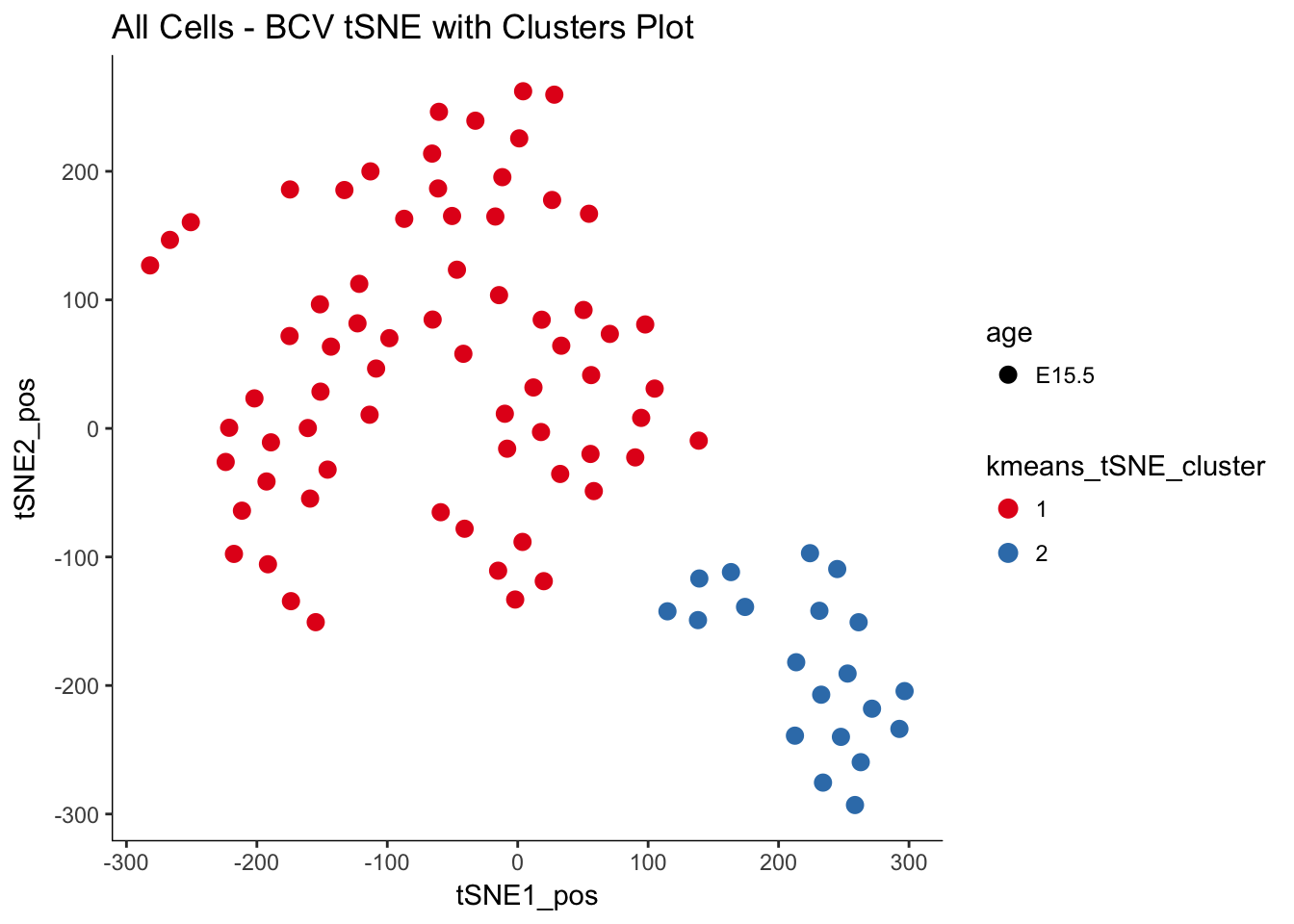
QC on the clusters
# Distribution of number of genes expressed in clusters
q <- ggplot(pData(e15.Mb.dat.filter)) +
geom_density(aes(x=num_genes_expressed,fill=kmeans_tSNE_cluster),alpha=0.3) + scale_fill_brewer(palette="Set1") + facet_grid(.~age) + monocle:::monocle_theme_opts()
q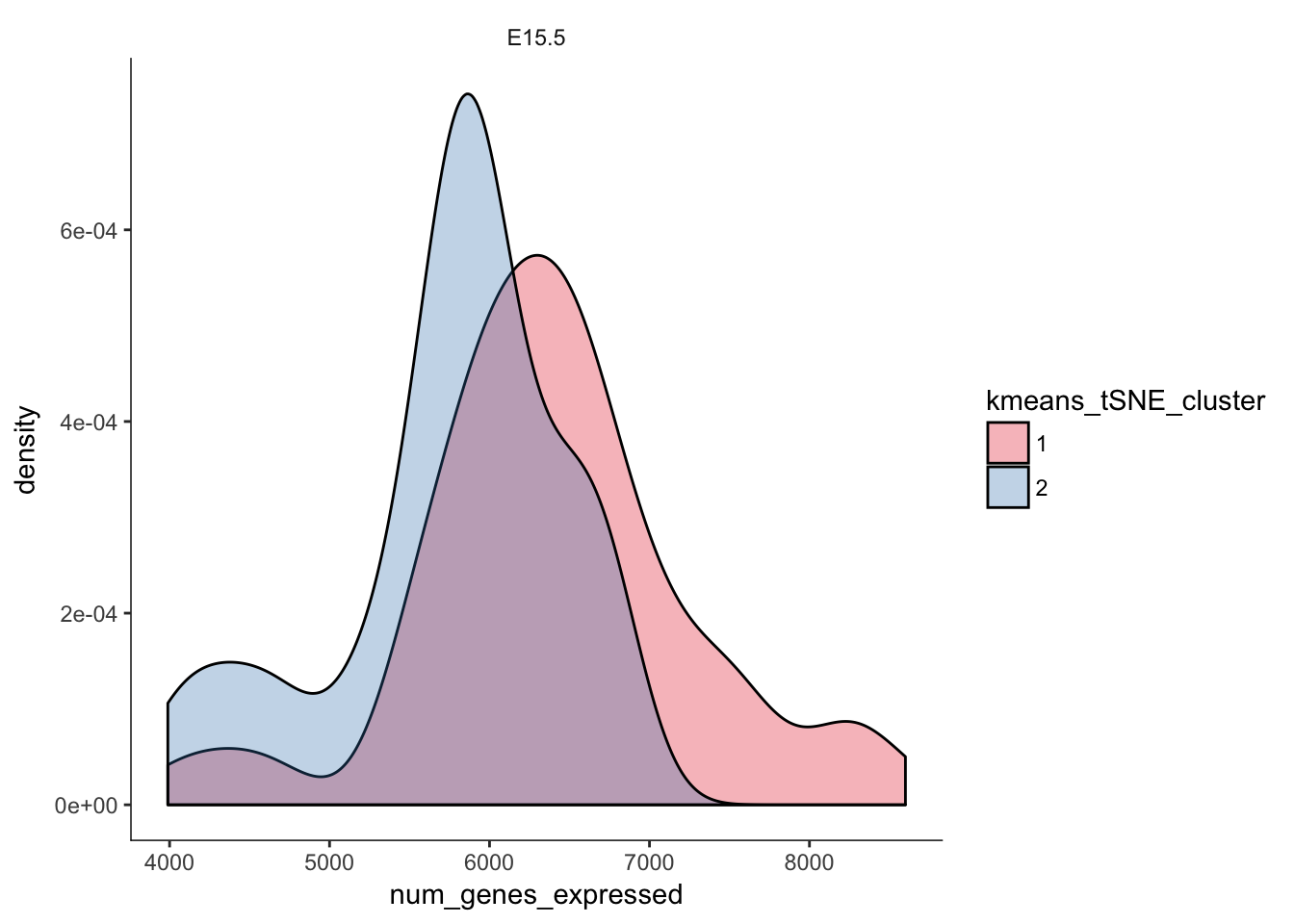
# Plotting the distribution of total mRNAs in clusters
q<-ggplot(pData(e15.Mb.dat.filter)) +
geom_density(aes(x=Total_mRNAs,fill=kmeans_tSNE_cluster),alpha=0.3) + scale_fill_brewer(palette="Set1") + facet_grid(.~age) + monocle:::monocle_theme_opts()
q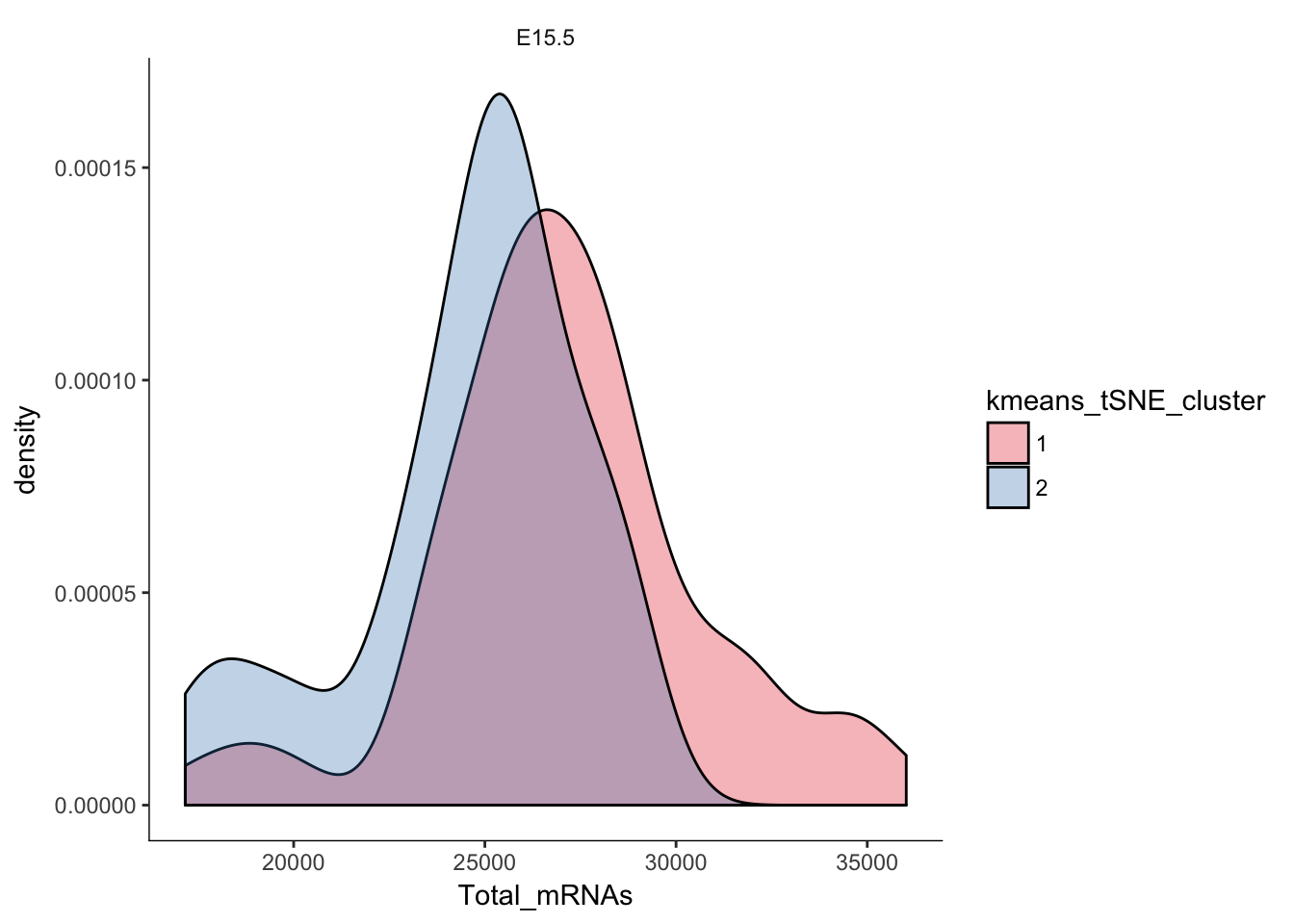
Saving the E15.5 MB final cds
e15.Mb.dat.filter.final <- e15.Mb.dat.filter
saveRDS(object = e15.Mb.dat.filter.final, file = file.path(Rdata_dir, "e15.Mb.dat.filter.final.Rds"))Extracting pData information and saving it
e15.Mb.clusters.df <- pData(e15.Mb.dat.filter)[,c(1,39)]
saveRDS(e15.Mb.clusters.df, file = file.path(Rdata_dir, "e15.Mb.clusters.df.rds"))Session Info
sessionInfo()## R version 3.3.0 (2016-05-03)
## Platform: x86_64-apple-darwin13.4.0 (64-bit)
## Running under: OS X 10.11.6 (El Capitan)
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] grid splines stats4 parallel stats graphics grDevices
## [8] utils datasets methods base
##
## other attached packages:
## [1] ADPclust_0.7 ggbiplot_0.55 scales_0.5.0
## [4] SC3_1.1.4 ROCR_1.0-7 jackstraw_1.1.1
## [7] lfa_1.2.2 tsne_0.1-3 gridExtra_2.3
## [10] slackr_1.4.2 vegan_2.4-4 permute_0.9-4
## [13] MASS_7.3-47 gplots_3.0.1 RColorBrewer_1.1-2
## [16] Hmisc_4.0-3 Formula_1.2-2 survival_2.41-3
## [19] lattice_0.20-35 Heatplus_2.18.0 Rtsne_0.13
## [22] pheatmap_1.0.8 tidyr_0.7.1 dplyr_0.7.4
## [25] plyr_1.8.4 heatmap.plus_1.3 stringr_1.2.0
## [28] marray_1.50.0 limma_3.28.21 reshape2_1.4.3
## [31] monocle_2.2.0 DDRTree_0.1.5 irlba_2.2.1
## [34] VGAM_1.0-2 ggplot2_2.2.1 Biobase_2.32.0
## [37] BiocGenerics_0.18.0 Matrix_1.2-11
##
## loaded via a namespace (and not attached):
## [1] RSelenium_1.7.1 colorspace_1.3-2 class_7.3-14
## [4] rprojroot_1.2 htmlTable_1.9 corpcor_1.6.9
## [7] base64enc_0.1-3 mvtnorm_1.0-6 codetools_0.2-15
## [10] doParallel_1.0.11 robustbase_0.92-7 knitr_1.17
## [13] jsonlite_1.5 cluster_2.0.6 semver_0.2.0
## [16] shiny_1.0.5 rrcov_1.4-3 httr_1.3.1
## [19] backports_1.1.1 assertthat_0.2.0 lazyeval_0.2.1
## [22] formatR_1.5 acepack_1.4.1 htmltools_0.3.6
## [25] tools_3.3.0 bindrcpp_0.2 igraph_1.1.2
## [28] gtable_0.2.0 glue_1.1.1 binman_0.1.0
## [31] doRNG_1.6.6 Rcpp_0.12.14 slam_0.1-37
## [34] gdata_2.18.0 nlme_3.1-131 iterators_1.0.8
## [37] mime_0.5 rngtools_1.2.4 gtools_3.5.0
## [40] WriteXLS_4.0.0 XML_3.98-1.9 DEoptimR_1.0-8
## [43] yaml_2.1.15 pkgmaker_0.22 rpart_4.1-11
## [46] fastICA_1.2-1 latticeExtra_0.6-28 stringi_1.1.5
## [49] pcaPP_1.9-72 foreach_1.4.3 e1071_1.6-8
## [52] checkmate_1.8.4 caTools_1.17.1 rlang_0.1.6
## [55] pkgconfig_2.0.1 matrixStats_0.52.2 bitops_1.0-6
## [58] qlcMatrix_0.9.5 evaluate_0.10.1 purrr_0.2.4
## [61] bindr_0.1 labeling_0.3 htmlwidgets_0.9
## [64] magrittr_1.5 R6_2.2.2 combinat_0.0-8
## [67] wdman_0.2.2 foreign_0.8-69 mgcv_1.8-22
## [70] nnet_7.3-12 tibble_1.3.4 KernSmooth_2.23-15
## [73] rmarkdown_1.8 data.table_1.10.4 HSMMSingleCell_0.106.2
## [76] digest_0.6.12 xtable_1.8-2 httpuv_1.3.5
## [79] openssl_0.9.7 munsell_0.4.3 registry_0.3This R Markdown site was created with workflowr